10.8 代谢组单组学快速分析示例
本章节将利用示例数据,完成一个代谢组单组学的从导入的完成基本常见的分析流程。
示例数据下载:1. Github下载地址 2. 百度网盘下载地址
10.8.1 代谢组数据导入
注意:
由于单一组学项目不涉及创建关系表,因此表达矩阵的样本名称必须与表型数据中样本名称完全一致。
由于单一组学项目不涉及创建关系表,因此表达矩阵的样本名称必须与表型数据中样本名称完全一致。
library(EasyMultiProfiler)
meta_data <- read.table('col.txt',header = T,row.names = 1)
data <- read.table('metabol.txt',header = T,quote = '',sep = '\t')
MAE <- EMP_easy_import(data = data,coldata = meta_data,
sampleID = rownames(meta_data),
type = 'normal')
10.8.2 代谢组数据查看
查看当前代谢组组学
MAE |>
EMP_assay_extract() # 查看表达矩阵
MAE |>
EMP_coldata_extract() # 查看表型数据
MAE |>
EMP_rowdata_extract() # 查看代谢物注释
10.8.3 构建质控的代谢物特征变异系数 (非必须)
根据表型数据的QC样本计算代谢物特征的变异系数
MAE |>
EMP_assay_extract() |>
EMP_mutate(CV = sd(c(QC1,QC2,QC3,QC4,QC5,QC6))/mean(c(QC1,QC2,QC3,QC4,QC5,QC6)),
mutate_by = 'feature',location = 'rowdata',action = 'rowwise',.before = 2)
根据变异系数进行代谢物筛选
MAE |>
EMP_assay_extract() |>
EMP_mutate(CV = sd(c(QC1,QC2,QC3,QC4,QC5,QC6))/mean(c(QC1,QC2,QC3,QC4,QC5,QC6)),
mutate_by = 'feature',location = 'rowdata',action = 'rowwise',.before = 2) |>
EMP_filter(feature_condition = CV < 0.3)
10.8.4 根据表型数据提取代谢物数据
根据表型数据中的MS2Metabolite列,将代谢物折叠成代谢物名称
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',
estimate_group = 'MS2Metabolite')
根据表型数据中的MS2kegg列,将代谢物折叠成KEGG ID
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',
estimate_group = 'MS2kegg')
10.8.5 代谢物ID转换
EMP包内置了代谢物在多个数据库之间进行ID转换,包括CAS,DTXSID,DTXCID,SID,CID,KEGG,ChEBI,HMDB,Drugbank。
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',
estimate_group = 'MS2kegg') |>
EMP_feature_convert(from = 'KEGG',to = 'HMDB')
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',
estimate_group = 'MS2kegg') |>
EMP_feature_convert(from = 'KEGG',to = 'Drugbank')
10.8.6 代谢物丰度批次矫正 (非必须)
MAE |>
EMP_assay_extract() |>
EMP_adjust_abundance(.factor_unwanted = 'Region',
.factor_of_interest = 'Group',
method = 'combat_seq')
10.8.7 代谢物数据标准化
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_decostand(method = 'relative')
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_decostand(method = 'clr')
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_decostand(method = 'log2')
10.8.8 代谢物差异分析
利用wilcox算法进行差异分析
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_diff_analysis(method = 'wilcox.test',estimate_group = 'Group') |>
EMP_filter(feature_condition = pvalue < 0.05)
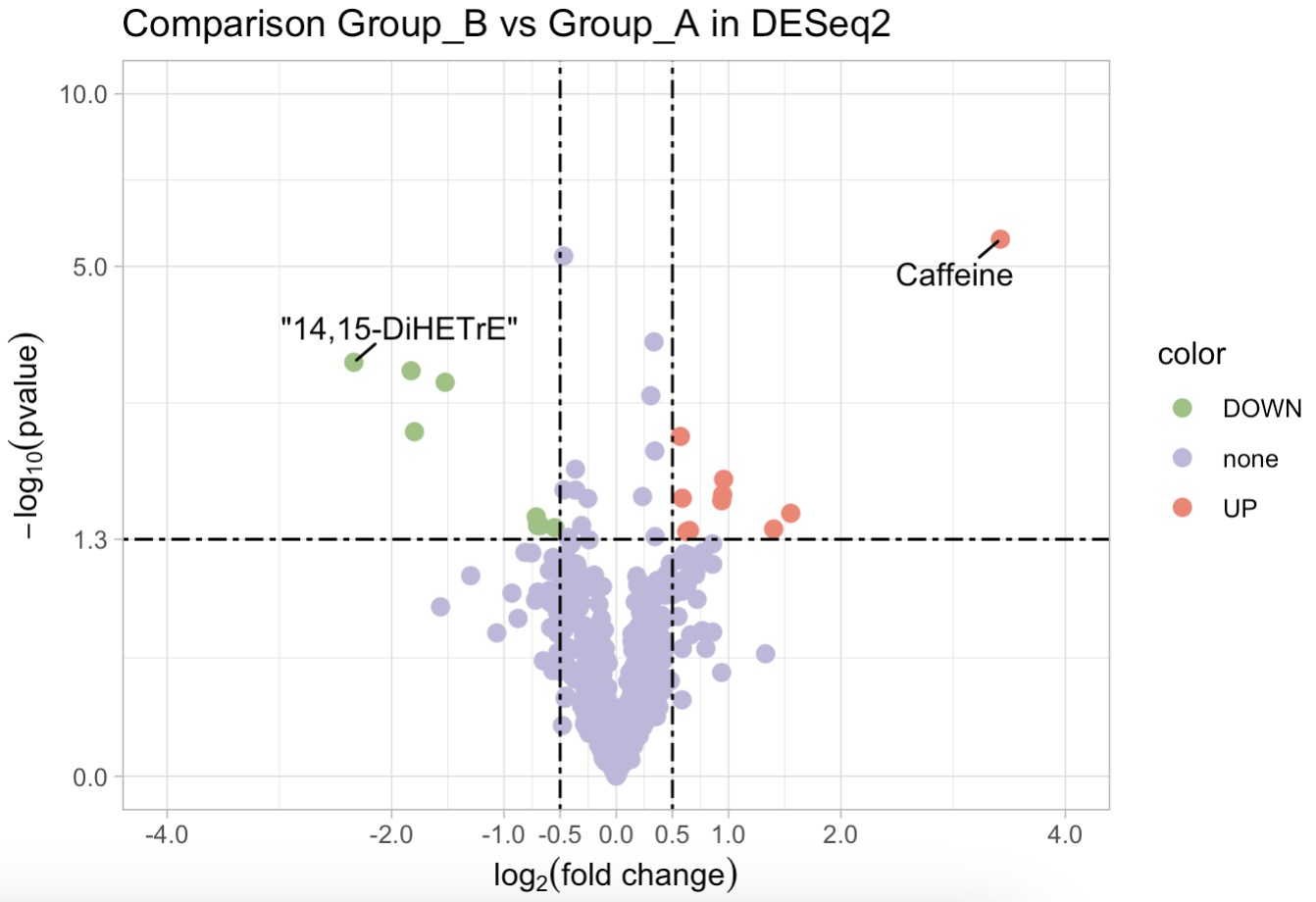
利用DESeq2算法筛选差异代谢物,并绘制火山图
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group) |>
EMP_volcanol_plot(key_feature = c('Caffeine','\"14,15-DiHETrE\"'),
palette = c('#FA7F6F','#96C47D','#BEB8DC'),
dot_size = 2.5,threshold_x = 0.5,mytheme = "theme_light()",
min.segment.length = 0, seed = 42, box.padding = 0.5)
10.8.9 构建PLS或OPLS降维模型
获取代谢物VIP值
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_dimension_analysis(method = 'pls',estimate_group = 'Group')
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_dimension_analysis(method = 'opls',estimate_group = 'Group')
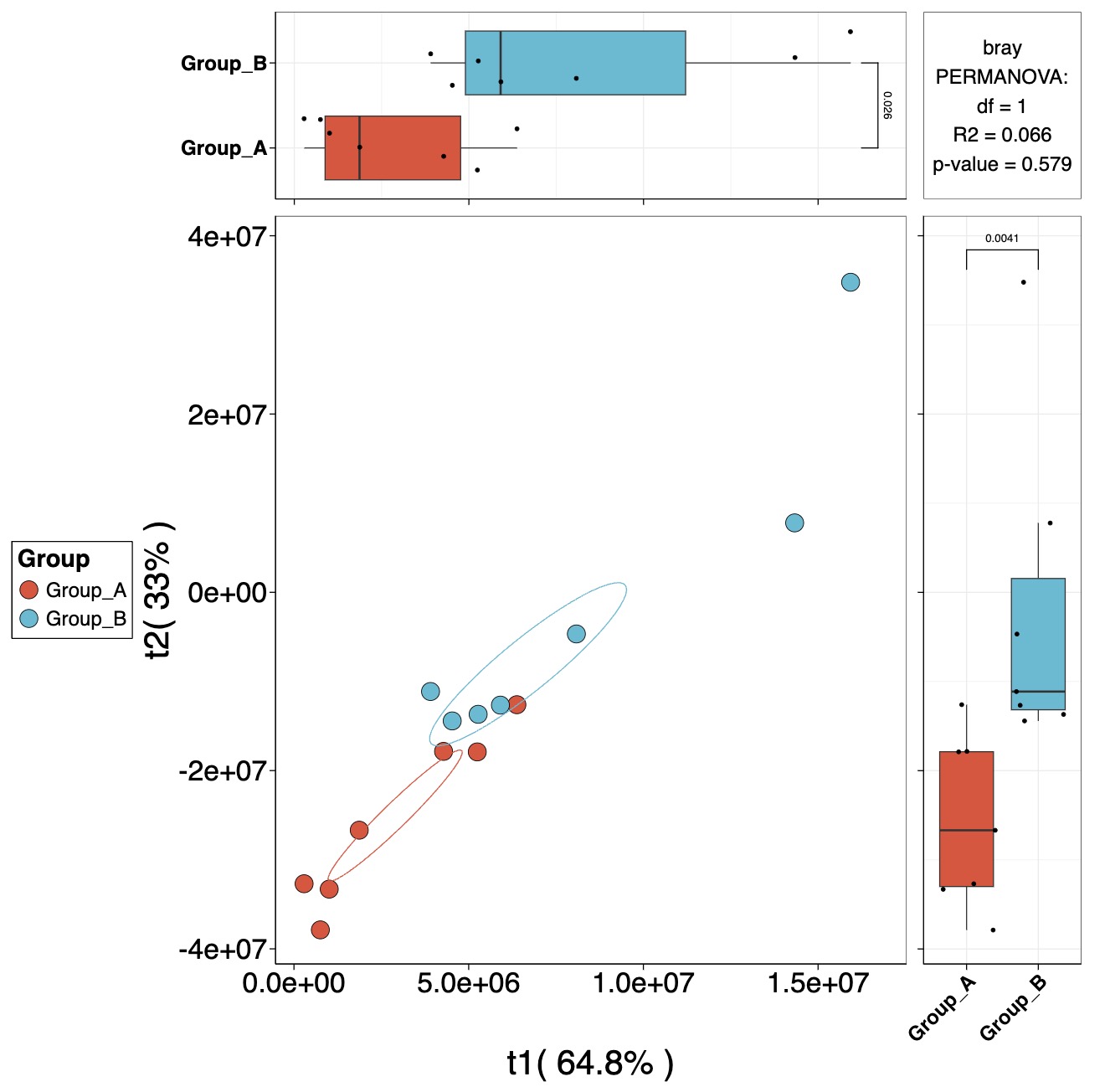
根据降维模型进行绘图 PLS降维
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_dimension_analysis(method = 'pls',estimate_group = 'Group') |>
EMP_scatterplot(show='p12html',ellipse=0.3)
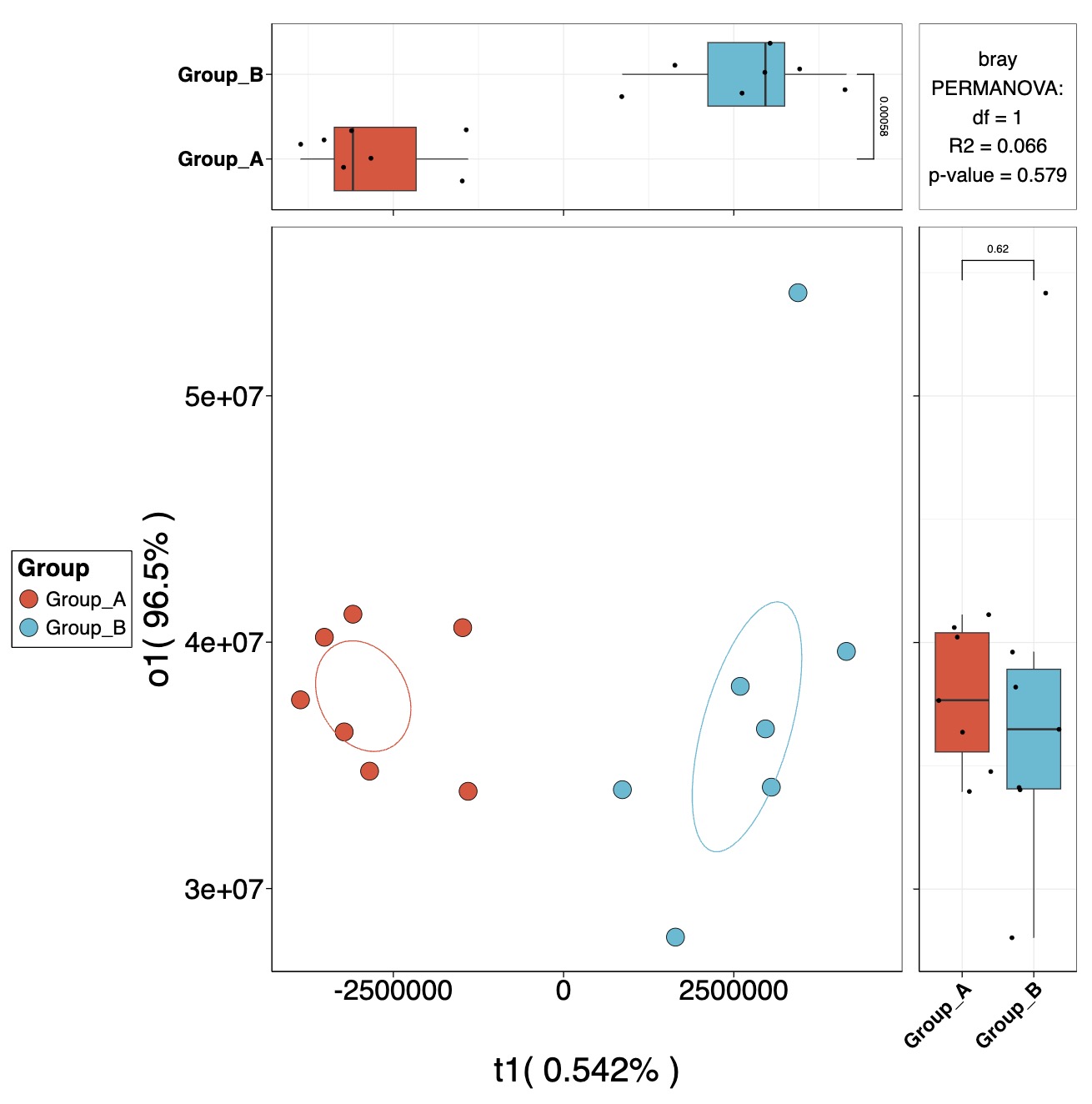
OPLS降维
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_dmension_analysis(method = 'opls',estimate_group = 'Group') |>
EMP_scatterplot(show='p12html',ellipse=0.3)
10.8.10 筛选关键代谢物
利用PLS模型和差异分析P值筛选代谢物
# 利用差异分析和降维分析的结果组合进行筛选,具体标准可以根据实际需要调整
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_diff_analysis(method = 'wilcox.test',estimate_group = 'Group') |>
EMP_dimension_analysis(method = 'pls',estimate_group = 'Group') |>
EMP_filter(feature_condition = VIP >1 & pvalue < 0.05 & abs(fold_change >1.2))
10.8.11 机器学习模型筛选代谢物
EMP包内置了Boruta算法、随机森林算法、xgboost算法和Lasso算法进行特征筛选。 详细用法可以使用help(EMP_marker_analysis)查看更多示例。
利用Boruta算法筛选关键代谢物并绘制热图
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2Metabolite') |>
EMP_marker_analysis(method = 'boruta',estimate_group = 'Group') |>
EMP_filter(feature_condition = Boruta_decision!= 'Rejected') |>
EMP_heatmap_plot(palette='Spectral',legend_bar='auto',
scale='standardize',
clust_row=TRUE,clust_col=TRUE)
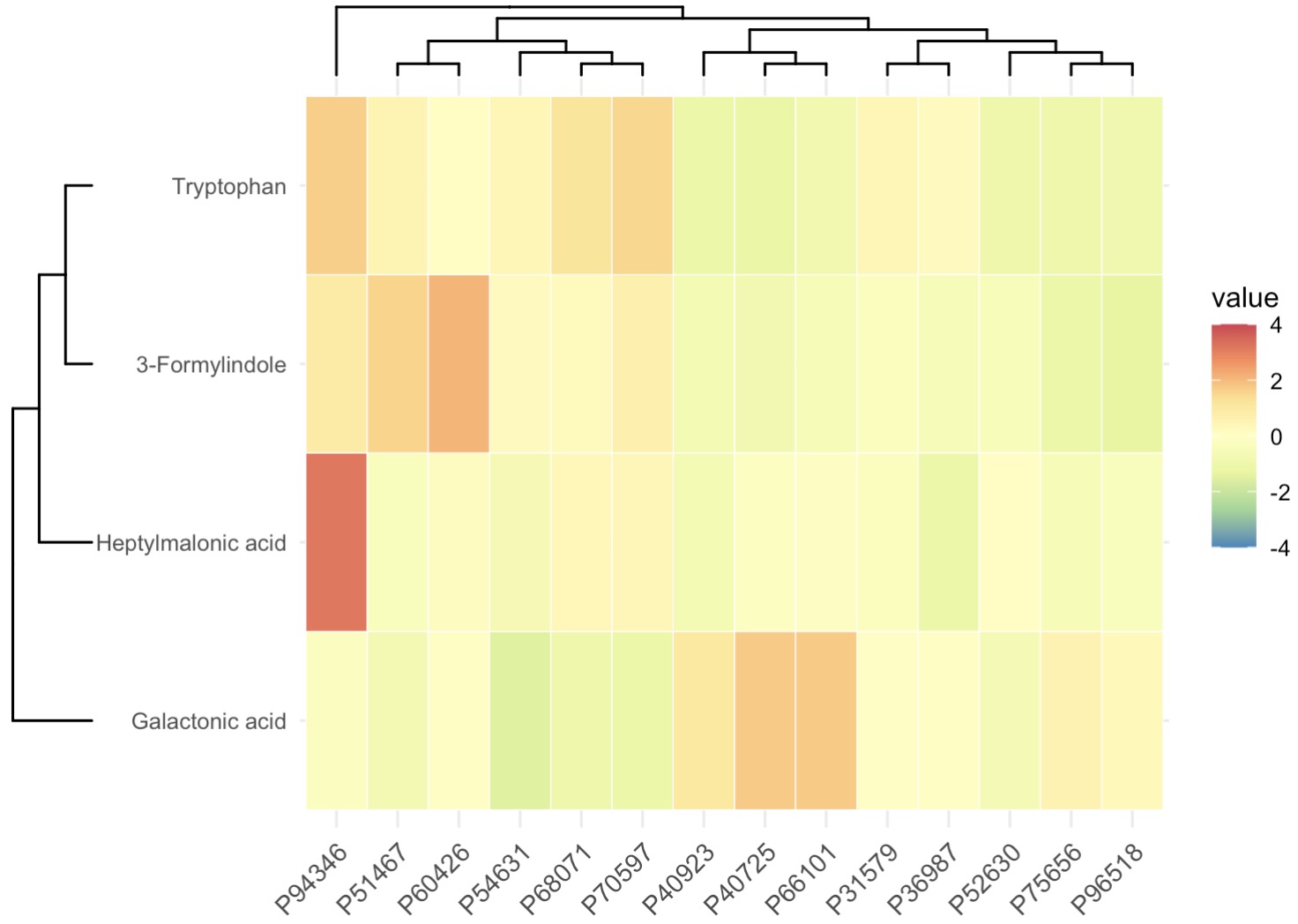
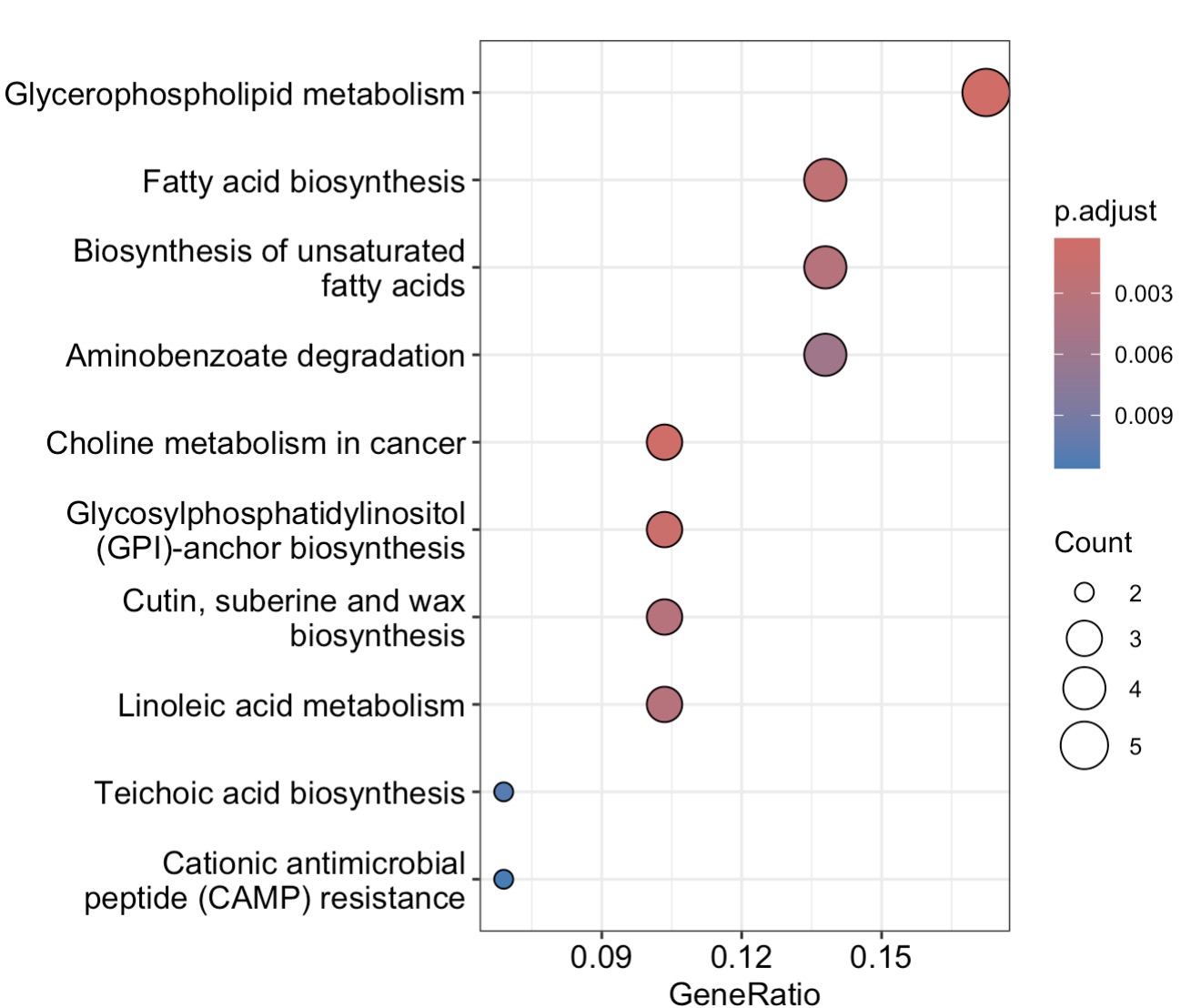
10.8.12 代谢组学KEGG富集方法
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2kegg') |>
EMP_dimension_analysis(method = 'pls',estimate_group = 'Group') |>
EMP_filter(feature_condition = VIP >1) |>
EMP_enrich_analysis( keyType ='cpd',
KEGG_Type = 'KEGG',
pvalueCutoff=0.05) |>
EMP_enrich_dotplot()
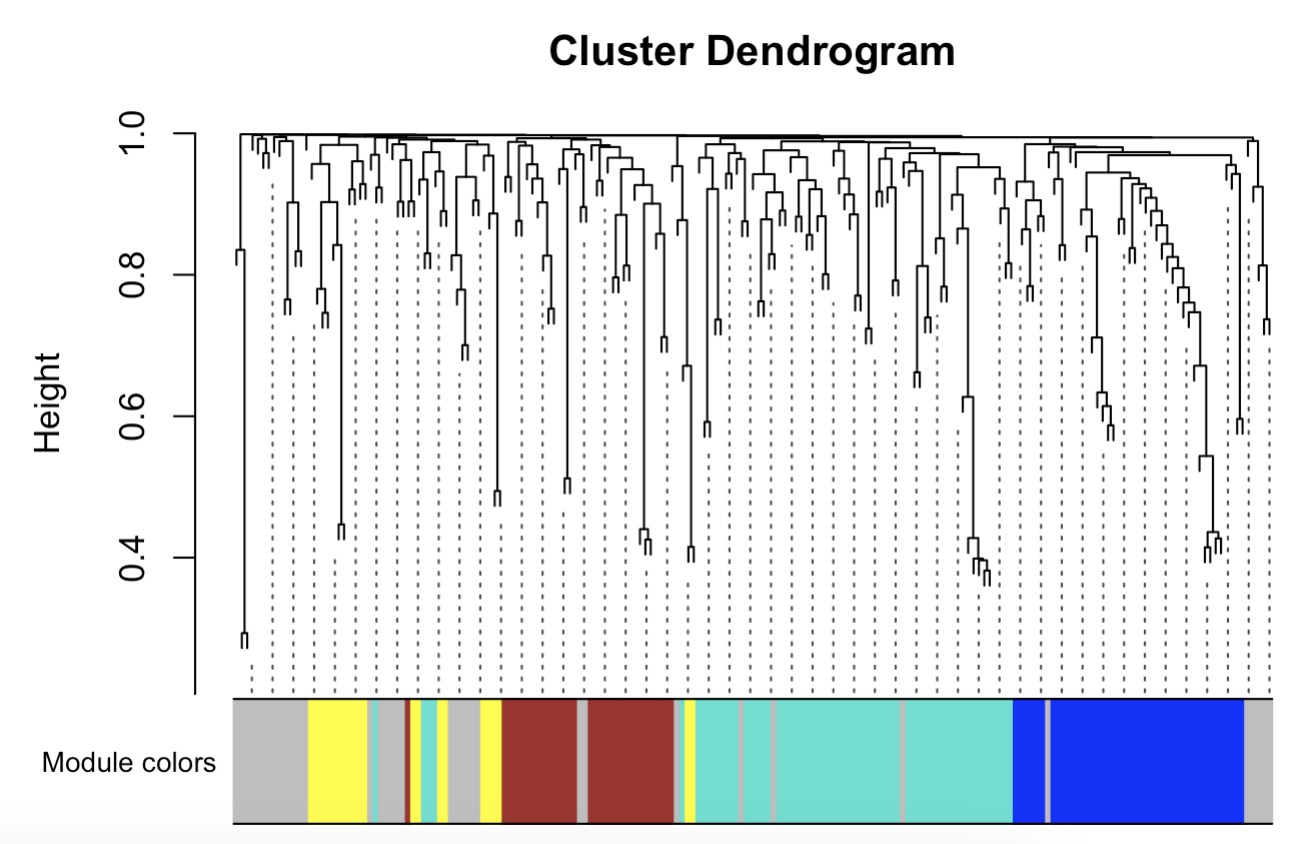
10.8.13 代谢组学WGCNA分析
第一步:根据表型数据获取代谢物聚类情况
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2kegg') |>
EMP_WGCNA_cluster_analysis(RsquaredCut = 0.8,
mergeCutHeight=0.2,
minModuleSize=10)
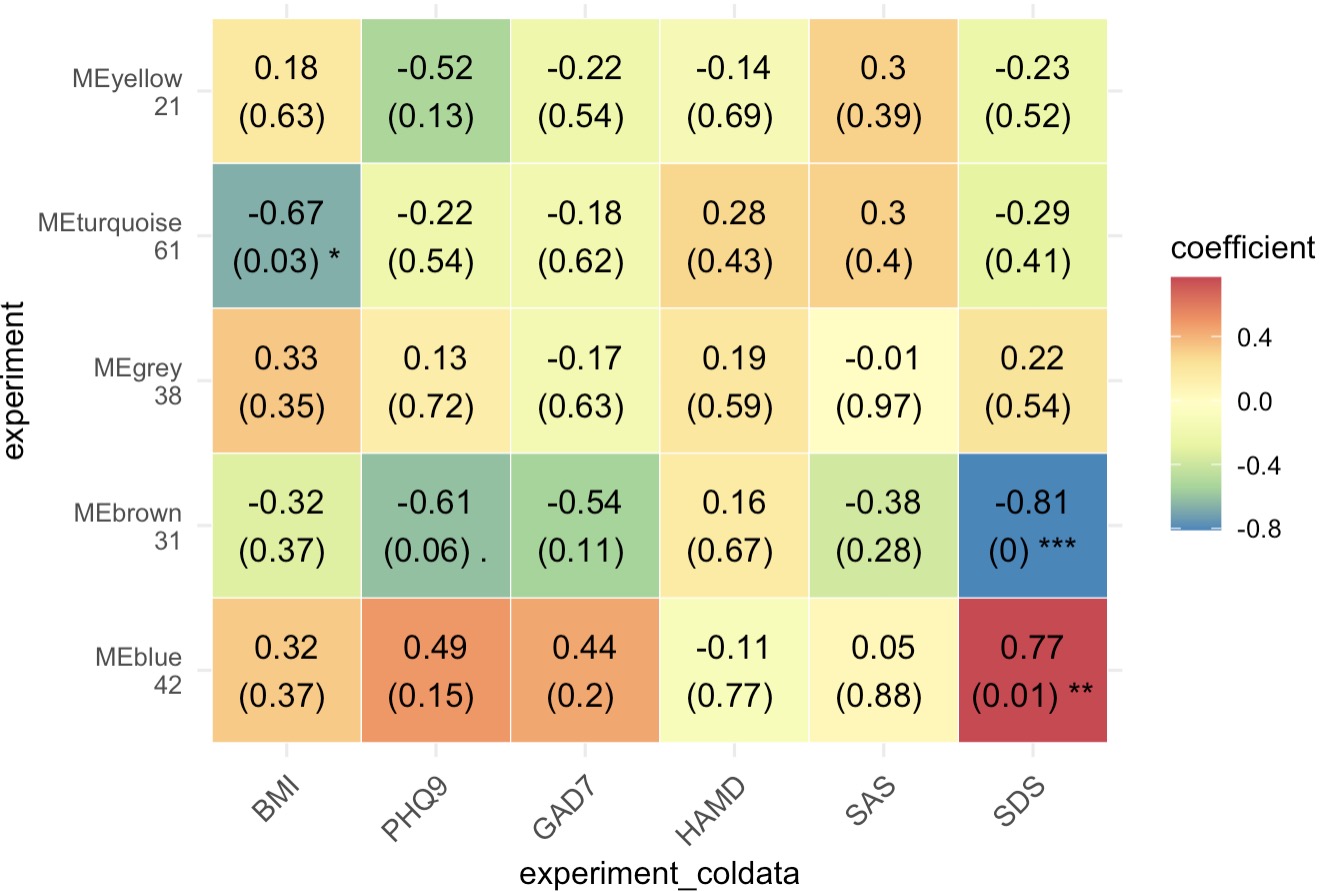
第二步:根据表型数据获取相关代谢簇热图
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2kegg') |>
EMP_WGCNA_cluster_analysis(RsquaredCut = 0.8,
mergeCutHeight=0.2,
minModuleSize=10) |>
EMP_WGCNA_cor_analysis(coldata_to_assay = c('BMI','PHQ9','GAD7','HAMD','SAS','SDS'),
method='spearman') |>
EMP_heatmap_plot(palette = 'Spectral')
第三步:筛选相关基因簇进行富集分析
MAE |>
EMP_assay_extract() |>
EMP_collapse(collapse_by = 'row',estimate_group = 'MS2kegg') |>
EMP_WGCNA_cluster_analysis(RsquaredCut = 0.8,
mergeCutHeight=0.2,
minModuleSize=10) |>
EMP_WGCNA_cor_analysis(coldata_to_assay = c('BMI','PHQ9','GAD7','HAMD','SAS','SDS'),
method='spearman') |>
EMP_heatmap_plot(palette = 'Spectral') |>
EMP_filter(feature_condition = WGCNA_color == 'blue' ) |>
EMP_enrich_analysis(keyType = 'cpd',KEGG_Type = 'KEGG') |>
EMP_enrich_dotplot()